uveiti infiammazioni dell'occhio - sintomi e trattamenti
La parola uveite è un termine descrittivo che definisce l'infiammazione intraoculare dell'uvea.
Questo termine viene comunemente utilizzato per definire anche infiammazioni che riguardano altre strutture oculari oltre all'uvea vera e propria (sclera, cornea, retina ecc) ma che vengono incluse nelle "uveiti".
Introduzione
Per eseguirlo ogni oculista segue un proprio iter mentale che tuttavia si basa su dei principi uguali per tutti:
- Anamnesi Generale
- Visita ed Anamnesi Oculistica
- Ipotesi diagnostica
- Richiesta di Esami Diagnostici
- Terapia
- Rivalutazione Diagnostica e Terapeutica
Anamnesi Generale:
L'anamnesi generale deve essere accuratissima ed allo scopo di non dimenticare alcun dettaglio vengono proposti dei questionari molto dettagliati che il paziente dovrebbe compilare prima della visita oculistica.Questa accuratezza, che allo stesso paziente può sembrare eccessiva, è invece indispensabile per poter evidenziare sintomi o segni di patologie d'organo correlabili all'uveite.
Visita ed Anamnesi Oculistica
La visita oculistica inizia con la raccolta dell'anamnesi oculare completa e sulla dettagliata ricostruzione storica dell'episodio uveitico e di eventuali altri pregressi problemi oculistici.I sintomi principali di una uveite sono variabili come intensità e durata in funzione della sede e dell'intensità dell'infiammazione. I sintomi più importanti sono il dolore, la fotofobia, la lacrimazione, il calo dell'acuità visiva e la visione di corpi mobili vitreali.
Dopo la valutazione della massima acuità visiva, si prosegue con un attento esame obiettivo alla lampada a fessura che consentirà di raccogliere dati importanti sulle strutture oculari esterne (congiuntiva, cornea e sclera) eventualmente coinvolte dall'infiammazione.
Congiuntiva: La presenza di "rossore congiuntivale" deve essere distinta tra l'iperemia congiuntivale superficiale tipica delle congiuntiviti (fini vasi congiuntivali dilatati) dall'iniezione pericheratica determinata dalla vasodilatazione dei vasi profondi (violacei) caratteristica delle uveiti.
Cornea: La cornea può essere la sede di partenza di infezioni acute e croniche dell'occhio (cheratiti, cheratouveiti ecc.) e/o la sede in cui i prodotti dell'infiammazione intraoculare, precipitati cheratici, si vanno a depositare.
La distribuzione, le dimensioni, l'età di questi precipitati corneali danno numerose informazioni utili all'inquadramento diagnostico.
Sclera: Il guscio sclerale può essere partenza di processi infiammatori gravi che lo colpiscono direttamente (sclerite) o meno gravi che interessano la membrana episclerale che lo ricopre (episclerite).
Successivamente si valutano, sempre alla lampada a fessura, le strutture interne della parte anteriore dell'occhio (camera anteriore, iride e cristallino).
Camera anteriore: L'uveite determina in camera anteriore la presenza di prodotti dell'infiammazione, cellule e proteine, che vanno quantificate durante ogni visita allo scopo di graduare lo stato dell'infiammazione.
Iride: L'iride è spesso sede di processi infiammatori granulomatosi (noduli di Koeppe e di Busacca), di esiti di infezioni o infiammazioni (atrofia o eterocromia) e/o di sinechie post-infiammatorie con il cristallino.
Cristallino: Il cristallino può presentare opacità legate alle caratteristiche specifiche di quella forma di uveite o causate dalla terapia cronica con steroide topico o sistemico.
Sempre alla lampada a fessura ed utilizzando delle lenti apposite, dopo dilatazione massima della pupilla con gocce midriatiche, si valutano le strutture interne oculari (corpi ciliari e pars plana, vitreo, coroide, retina, vasi retinici e papilla ottica).
Corpi Ciliari e Pars Plana: Questa è la sede dell'uveite intermedia ed è valutabile solo con pupilla dilatata e con una lente speciale alla lampada a fessura. Si possono apprezzare, a livello vitreale, degli essudati rotondi (snow ball) o placoidi (snow banks) e dei segni di vasculite retinica periferica.
Vitreo: La comparsa nel gel vitreale di cellule infiammatorie, di briglie fibrose ed essudazioni può essere primitiva (vitreite) o secondaria ad un'infiammazione del segmento posteriore dell'occhio.
Coroide: L'infiammazione della coroide (coroidite) è caratterizzata da chiazze giallastre sottoretiniche a margini netti. La retina soprastante la lesione può rimanere indenne o partecipare in un secondo tempo all'infiammazione (corioretinite).
Retina: L'infiammazione retinica (la retinite) è caratterizzata da un'area di sbiancamento retinico a margini non ben definiti con vasculite retinica associata.
Se l'infiammazione interessa anche la coroide si parlerà di retinocoroidite. Una grave forma di uveite essudativa può portare al distacco di retina con grave compromissione della vista.
In altre forme di uveite posteriore si ha invece la formazione secondaria di neovasi sottoretinici maculari che vanno a complicare patologie infiammatorie croniche.
Vasi Retinici: La vasculite è l'infiammazione delle pareti vascolari del circolo retinico sia venoso (flebite) che arterioso (arterite). I vasi si presentano lungo il loro decorso delle infiltrazioni biancastre.
La vasculite può presentarsi da sola o secondariamente a patologie della retina e della coroide.
Papilla Ottica: La papilla ottica può essere interessata direttamente da un processo infiammatorio (papillite) o da una sua infiltrazione di tessuto granulomatoso.
Altre alterazioni della papilla ottica possono essere secondarie ad ipotonia bulbare post-uveitica (edema della papilla ottica), ad un papillare diretto (atrofia della papilla ottica).
Ipotesi diagnostiche:
 L'oculista dopo aver eseguito l'anamnesi generale, l'anamnesi oculare e la valutazione clinica del paziente, ha in mano un
insieme di dati preliminari importanti a cui devono essere rielaborati ed incrociati tra di loro allo scopo di cominciare a
formulare delle ipotesi diagnostiche.
L'oculista dopo aver eseguito l'anamnesi generale, l'anamnesi oculare e la valutazione clinica del paziente, ha in mano un
insieme di dati preliminari importanti a cui devono essere rielaborati ed incrociati tra di loro allo scopo di cominciare a
formulare delle ipotesi diagnostiche.Inizialmente bisogna cercare di classificare l'uveite in funzione dei parametri obiettivi ed anamnestici raccolti.
Le uveiti vengono classificate utilizzando dei parametri differenti: anatomici, clinici, etiologici o anatomopatologici.
Nessuno di questi soddisfa completamente le necessità d'inquadramento diagnostico ma ognuna di queste classificazioni consente all'oculista di perseguire la difficile arte della diagnosi differenziale.
Classificazione clinica: Sulla base dei tempi di insorgenza della malattia oculare l'uveite può essere suddivisa in acuta (primo episodio di durata inferiore alle 6 settimane) o cronica (durata superiore ai 6 mesi).
Inoltre possiamo definire la gravità dell'uveite come lieve, media o grave. Inoltre dobbiamo valutare se l'uveite è monolaterale o bilaterale.
Classificazione patologica: La divisione clinica delle uveiti dal punto di vista patologico in granulomatose e non granulomatose non è semplice e si basa sul rispetto di alcuni parametri clinico anamnestici.
La forma granulomatosa è in genere un uveite che inizia in maniera insidiosa ed è tendenzialmente cronica.
Anatomicamente le lesioni caratteristiche sono i precipitati corneali a grasso di montone e l'eventuale presenza di lesioni nodulari al segmento posteriore dell'occhio.
Le forme non granulomatose sono in genere uveiti acute, di breve durata caratterizzate soprattutto da dolore e rossore oculare. I precipitati corneali sono piccoli e l'interessamento al segmento posteriore è diffuso.
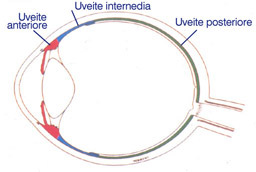
Classificazione anatomica: Le uveiti, da un punto di vista anatomico, vengono divise in tre gruppi principali in funzione della parte anatomica interessata dall'infiammazione o infezione che sia.
Distinguiamo quindi le uveiti anteriori, le intermedie e le posteriori.
Le uveiti sono definite "anteriori" quando si ha l'interessamento patologico del segmento anteriore dell'occhio (cornea, iride, corpi ciliari). In particolare si può definire irite se colpisce solo l'iride ed irido ciclite quando vi è un interessamento dell'iride e dei corpi ciliari.
La cheratite riguarda invece solo l'infiammazione della cornea.
Le uveiti vengono definite "intermedie" quando l'infiammazione colpisce la parte posteriore dei corpi ciliari e l'estrema periferia retinica, detta pars plana.
Le uveiti sono definite "posteriori" quando l'infiammazione è localizzata nel segmento posteriore dell'occhio.
In particolare a secondo della localizzazione iniziale dell'infezione si può parlare di retinite, retinocoroidite, corioretinite o coroidite.
Infine si parla di "panuveite" quando la flogosi interessa contemporaneamente tutti i segmenti del bulbo oculare.
Classificazione etiologica: La prima fondamentale suddivisione da eseguire è tra "uveite infettiva" e "uveite non infettiva".
Questa prima e difficile suddivisione è determinante per la scelta terapeutica.
Le forme infettive sono secondarie all'impianto di germi dall'esterno (forme esogene) o dall'interno (forme endogene). Queste ultime possono essere secondarie ad infezioni sistemiche o locali di virus, batteri, funghi e parassiti.
Tra le forme non infettive (forme endogene) esistono uveiti secondarie ad artriti e malattie granulomatose.
Altre forme di uveiti non infettive riguardano le forme "idiopatiche" sia quelle inquadrabili in una sindrome specifica sia quelle non inquadrabili in nessuna sindrome (25% del totale).
Richiesta di esami strumentali:
Gli esami strumentali ed ematologici verranno richiesti in maniera mirata e molto selettiva sia per motivi economici sia perché una pletora di esami inutili può fuorviare la diagnosi differenziale.Bisogna sempre ricordare che lo scopo per cui si richiedono ulteriori esami strumentali è quello di avvalorare o meno le ipotesi diagnostiche precedentemente formulate.
Gli esami vengono anche richiesti allo scopo di valutare la fattibilità di alcune terapie sistemiche.
Gli esami minimi iniziali sono: Emocromo con formula, Funzionalità epatica, Funzionalità renale, VES.
A questi vanno aggiunti esami più specifici che la storia clinica del paziente e la valutazione oculistica indicano come utili approfondimenti (ANA, Ac. Anticardiolipina, ANCA, Fattore reumatoide, ACE, Tipizzazione HLA totale, Test cutanei per TBC, Biopsie congiuntivali, Biopsie ghiandole lacrimali, PCR su acqueo e vitreo, RX torace RX articolazioni sacro iliache, Scintigrafia al Gallio ecc.).
Altri esami di approfondimento e documentazione riguardano l'aspetto prettamente oculistico della patologia (Fluorangiografia retinica, Angiografia retinica con verde indocianina, Tomografia ottica a radiazione Coerente, Ecografia oculare, Perimetria computerizzata ecc.).
Quindi l'oculista personalizzerà le richieste di accertamenti sulla base di una prima classificazione eseguita (manifestazioni iniziali di uveite, uveite anteriore ricorrente, uveite intermedia, uveite posteriore, retinite, vasculite, sclerite) e solo successivamente ne richiederà degli altri se necessari.
Terapia:
Se il paziente è acuto o con grave rischio di perdita di visus la terapia deve essere iniziata immediatamente.Mentre nel caso di forme croniche o a lenta evoluzione si possono attendere i risultati delle indagini strumentali o degli esami ematologici iniziali.
Prima di iniziare ogni trattamento bisogna porsi l'obiettivo da raggiungere ed il tempo in cui si pensa ragionevolmente di ottenerlo.
Gli obiettivi possono essere così riassunti: Guarigione completa, scomparsa dall'infiammazione, terapia o profilassi dell'EMC, riduzione del numero delle recidive (profilassi). Un altro parametro da valutare è la necessità del trattamento che si basa sul grado di infiammazione, sulla localizzazione della lesione, sull'acuità visiva e sulla prognosi.
La scelta di eseguire immediatamente o no un trattamento dipenderà dal grado di infiammazione, dal grado di infiammazione.
La scelta della terapia si basa sulla precedente terapia eseguita, sul numero di recidive, sulla mono o bilateralità delle lesioni, sull'età del paziente e sulla sua compliance, sulla possibilità di poter seguire il paziente e sugli effetti collaterali dei farmaci da usare.
Rivalutazione diagnostica e terapeutica:
La rivalutazione del paziente e la rimessa in discussione della diagnosi iniziale è un aspetto importante dell'approccio all'uveite.Una critica lettura degli esami richiesti, dei risultati terapeutici ottenuti, dell'ipotesi diagnostica iniziale devono essere sempre eseguiti ad ogni visita di controllo senza essere condizionati da diagnosi precedentemente eseguite.
Tumori Oculari
Il contenuto e le informazioni della presente pagina sono integralmente derivate dal sito www.uveiti.it a cura del dott. Giulio Modorati - Responsabile del Servizio di Oncologia ed Immunopatologia Oculare dell' Ospedale San Raffaele di Milanoscarica il file : Tumori Oculari Benigni
Uveiti posteriori
Questo gruppo di uveiti si manifesta con lesioni della coroide e del complesso EPR-coriocapillare.Alcune sono patologie infiammatorie, altre patologie infettive e quasi sempre associate ad altre manifestazioni oculari ed extraoculari.
I processi infiammatori che interessano la retina difficilmente rimangono localizzati nel tessuto primariamente affetto ma diffondono nelle strutture vicine determinando unuveo-retinite.
Si parla pertanto di retino-coroidite quando una primitiva retinite interessa secondariamente la sottostante coroide e viceversa di corio-retinite quando una primitiva flogosi della coroide interessa secondariamente la retina.
Secondo la più attuale classificazione proposta dal SUN le uveiti posteriori possono essere divise in:
- Coroiditi focali, multifocali, diffuse
- Corioretiniti
- Retinocoroiditi
- Retiniti
- Neuroretiniti
Un modo schematico per approcciarsi alla diagnosi di uveite posteriore può essere la suddivisione delle manifestazioni cliniche oculari in base alla localizzazione della lesione o dellinfiammazione:
- U. posteriori con coinvolgimento predominante del vitreo
- U. posteriori con coinvolgimento predominante della retina superficiale
- U. posteriori con coinvolgimento predominante della retina profonda e/o della coroide con aspetto focale o multifocale
- Neuroretiniti
1. Uveiti posteriori con coinvolgimento predominante del vitreo
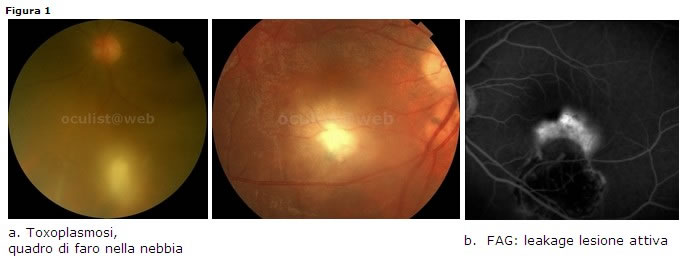
Toxoplasmosi
 La malattia può essere congenita (trasmissione transplacentare) con localizzazione oculare solitamente bilaterale o
acquisita per ingestione di oocisti o cisti tessutali (trasmissione oro-fecale).
La malattia può essere congenita (trasmissione transplacentare) con localizzazione oculare solitamente bilaterale o
acquisita per ingestione di oocisti o cisti tessutali (trasmissione oro-fecale).
Clinica: la reazione cellulare vitreale sovrastante il focolaio retinico (solitamente satellite di una lesione cicatriziale) può essere molto marcata (quadro di faro nella nebbia) (Fig. 1a). Lassenza di cellule infiammatorie nel vitreo posteriore è sempre un elemento di esclusione nella diagnosi di retinocoroidite toxoplasmica in fase attiva.
Diagnosi:
- Sierologia con valore limitato, possibile la ricerca anticorpi in umor acqueo (elevata sensibilità) e/o limpiego della PCR (alta specificità).
- FAG: leakage a livello del focolaio attivo, ipofluorescenze satelliti (75%) della lesione attiva (Fig. 1b).
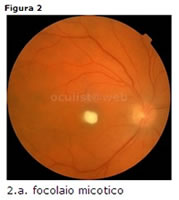
Candidosi
 Tossicodipendenza, AIDS, terapia sistemica prolungata, nutrizione parenterale sono le condizioni più comuni che predispongono ad infezioni micotiche opportunistiche.
Tossicodipendenza, AIDS, terapia sistemica prolungata, nutrizione parenterale sono le condizioni più comuni che predispongono ad infezioni micotiche opportunistiche. Infezioni esogene possono svilupparsi dopo chirurgia intraoculare ma più rare e meno aggressive rispetto alle forme endogene [32].
Clinica
monolaterale/bilaterale, lesioni bianco-giallastre cotonose, che attraversano la limitante interna e invadono la camera vitrea. Inizialmente i mezzi diottrici sono limpidi ma levoluzione verso lendoftalmite è la regola (Fig. 2).Diagnosi
prevalentemente anamnestico-clinica; vitrectomia diagnostica con striscio e/o coltura vitreale.Toxocariasi
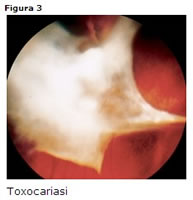 Colpisce soprattutto giovani con un range compreso tra i 2 e i 30 anni [33].
Colpisce soprattutto giovani con un range compreso tra i 2 e i 30 anni [33].Clinica:
endoftalmite, granuloma corioretinico maculare o posteriore; granuloma corioretinico periferico con trazioni vitreo-retiniche (Fig. 3).
Diagnosi: Sierologia (titolo positivo >1:32), aumento IgE, ipereosinofilia, ricerca anticorpi in UA/vitreo con test ELISA [34].
Sindrome di Whipple
E caratterizzata da coinvolgimento intestinale e neurologico, artrite monoarticolare e linfadenopatia.Clinica: opacità vitreali (tipo more bianche), vasculite e papilledema.
Diagnosi:
- VTK diagnostica,
- biopsia intestinale.
Malattia di Behçet
 Colpisce prevalentemente giovani adulti tra la 3°-4° decade; M>F; più diffusa nel bacino del Mediterraneo e Giappone.
Colpisce prevalentemente giovani adulti tra la 3°-4° decade; M>F; più diffusa nel bacino del Mediterraneo e Giappone.
Clinica
afte orali (98%), afte genitali, disturbi articolari, disturbi vascolari (tromboflebiti, occlusioni), disturbi neurologici. Le manifestazioni oculari posteriori caratteristiche comprendono vasculite occlusiva (89%) venosa e arteriosa con emorragie ed essudati, coroidite, corioretinite. Può associarsi a diffusa vitreite (Fig. 4).Diagnosi
- HLA-B51
- FAG: vasculite retinica e leakage papillare e peripapillare (negli stadi precoci della malattia)
Retinite da Herpes (ARN, Acute Retinal Necrosis)
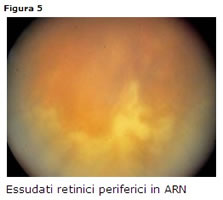 Quadro clinico drammatico che colpisce adulti con stato immunitario indifferente (interessa anche immunocompetenti), diversamente dalla PORN dove i pazienti sono quasi sempre immunodepressi [35].
Quadro clinico drammatico che colpisce adulti con stato immunitario indifferente (interessa anche immunocompetenti), diversamente dalla PORN dove i pazienti sono quasi sempre immunodepressi [35].
Clinica
focolai giallo-bianchi profondi a margini netti, tipicamente periferici negli stadi iniziali (Fig. 16) e che confluiscono progressivamente verso il polo posteriore; vasculite occlusiva essudativo-emorragica. Si può associare unimportante reazione infiammatoria granulomatosa in camera anteriore con ipertono ed una vitreite da moderata a severa.E frequente un distacco retinico essudativo dopo 4-6 settimane dallesordio della malattia.
Diagnosi
- gli esami supportano la diagnosi che è prevalentemente clinica.
- Utile la PCR sullumore acqueo per la ricerca di HZV, HSV 1-2 (CMV?, EBV?) [36].
- Test HIV.
- sierologia sifilide.
Retinite da CMV
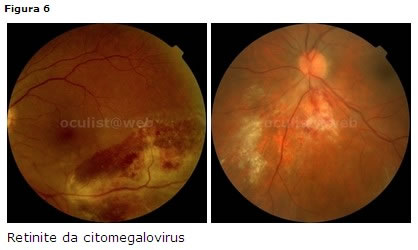 Colpisce soggetti immunodepressi (fattore di rischio linfociti T CD4 < 50 cell/mm³, non terapia HAART). Può essere la prima manifestazione di AIDS nel 2% dei casi [37].
Colpisce soggetti immunodepressi (fattore di rischio linfociti T CD4 < 50 cell/mm³, non terapia HAART). Può essere la prima manifestazione di AIDS nel 2% dei casi [37].
Clinica
inizia tipicamente con uno o due foci lungo i vasi con essudati intraretinici ed emorragie sul bordo della lesione. Si può associare a vasculite occlusiva mentre la reazione vitreale è modesta (Fig. 6).Levoluzione delle lesioni consiste in aree grigie inattive o cicatrici gliotiche trasparenti.
In pazienti che iniziano la HAART con preesistente retinite attiva, è possibile un fenomeno definito IRU (immune ricovery uveitis) ovvero una reazione infiammatoria importante che può essere un fenomeno transitorio o che può protrarsi con complicanze croniche.
Diagnosi
- test HIV,
- sierologia CMV.
In casi non responsivi alla terapia si esegue PCR su campioni biologici.
Sarcoidosi retinica
E possibile un esordio acuto (S. di Lofgren), subacuto, insidioso (S. di Heerfordt-Waldenstrom). Età 20-40 anni.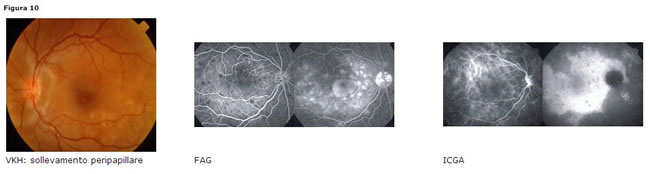
Clinica
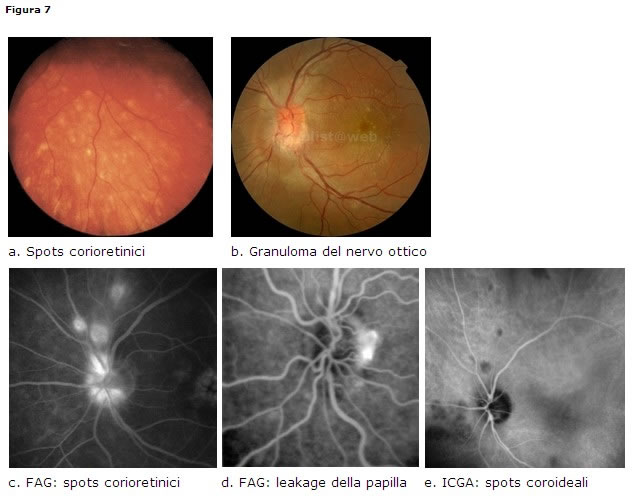
le lesioni del segmento posteriore, di solito monolaterali in fase acuta, colpiscono il 25% dei pazienti [38] con sarcoidosi oculare. Le manifestazioni a livello del segmento posteriore sono: vitreite, corio-retinite con spots bianchi rotondeggianti multipli inferiori, periflebite principalmente venosa a gocce di cera, occlusioni vascolari, neovascolarizzazione retinica, essudati vitreali basali a palla di neve o a collana di perle (pars planite), papilledema e granuloma della testa del nervo ottico (Fig. 7).Diagnosi
- ACE, lisozima, Mantoux, Rx torace, scintigrafia Ga67, biopsia del granuloma.
- FAG + ICGA: il verde indocianina è importante per identificare lesioni coroideali subcliniche che appaiono come spots ipofluorescenti nelle fasi intermedie e diventare iso o restare ipofluorescenti nelle fasi tardive.
TBC retinica
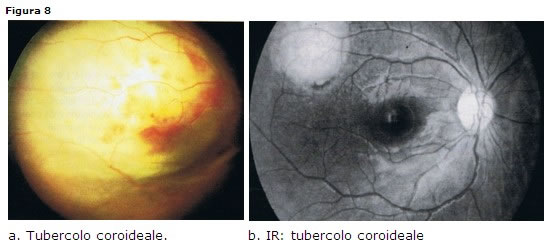 Colpisce prevalentemente immunodepressi e immigrati da aree endemiche. Interessa solo l1.4% dei pazienti con malattia in fase attiva.
Colpisce prevalentemente immunodepressi e immigrati da aree endemiche. Interessa solo l1.4% dei pazienti con malattia in fase attiva. Clinica
da vitreite di basso grado fino a vitreite importante con snowballs nel settore inferiore (pars planite); tubercoli coroideali singoli o multipli grigio-biancastri (Fig. 8 a-b) e periflebite sia periferica sia dei grossi vasi.Diagnosi
- PPD
- FAG + ICGA: lutilizzo del verde indocianina è importante per identificare lesioni subcliniche; le lesioni possono presentarsi come spots ipofluorescenti nelle fasi intermedie e diventare iso o restare ipofluorescenti nelle fasi tardive. Altri segni allICGA, indicanti una fase acuta, sono piccole lesioni iperfluorescenti dette pin-points e liperfluorescenza coroideale tardiva [39].
3. Coinvolgimento predominante della retina profonda e/o della coroide
Sifilide
Sono colpiti soggetti che hanno rapporti sessuali a rischio. Si presenta con lesioni cutanee (rash maculo-papuloso mani e piedi) e manifestazioni neurologiche (sifilide terziaria).Clinica
coroidite diffusa, corioretinite, vasculite (soprattutto periflebite) ed emorragie preretiniche. Può coesistere unimportante vitreite.Evolve in estese cicatrici corioretiniche con intensa migrazione pigmentaria.
Diagnosi
- Clinica
- VDRL e FTA-ABS
- Richerca HIV
- Esame liquor in HIV+
Corioretinopatia tipo birdshot
 Età media 50 anni, vitiligine.
Età media 50 anni, vitiligine.
Clinica
solitamente bilaterale, cellularità lieve o moderata in camera vitrea, spots corioretinici ovali color crema o depigmentati senza aree di iperpimentazione retroequatoriali, edema maculare cistoide, restringimento arteriolare, edema della testa del nervo ottico.Diagnosi
- HLA-A29 (sensibilità del 96% e specificità del 93%) [40].
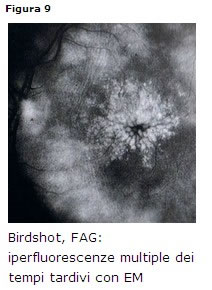
- FAG: si evidenzia unipofluorescenza precoce e uniperfluorescenza tardiva delle chiazzette (Fig. 9). E comune liperfluorescenza del nervo ottico.
- ICGA: lesioni ipofluorescenti precoci (per occlusione dei vasi arteriosi coroideali), iperfluorescenti nei tempi tardivi.
- ERG: anomali.
Sindrome di Vogt-Koyanagi-Harada
 Interessa soggetti intorno alla 3°-4° decade, più frequentemente giapponesi e ispano-americani e più spesso di sesso femminile. Le manifestazioni sistemiche sono: disturbi neurologici (atassia e confusione) e uditivi, poliosi delle ciglia, vitiligine, madarosi e alopecia negli stadi più avanzati.
Interessa soggetti intorno alla 3°-4° decade, più frequentemente giapponesi e ispano-americani e più spesso di sesso femminile. Le manifestazioni sistemiche sono: disturbi neurologici (atassia e confusione) e uditivi, poliosi delle ciglia, vitiligine, madarosi e alopecia negli stadi più avanzati. Clinica
coinvolgimento bilaterale, sollevamento corioretinico peripapillare (Fig. 10a), distacchi multipli sierosi retinici, edema della testa del nervo ottico, depigmentazione della coroide negli stadi più avanzati della malattia (fundus con aspetto a tramonto infuocato) con cicatrici corioretiniche multiple.Diagnosi
- FAG: precoci iperfluorescenze multiple a livello dellEPR, staining del fluido sottoretinico nelle fasi tardive
- ICGA: ipofluorescenza intermedia e iperfluorescenza tardiva
- Esame liquor cefalorachidiano (pleiocitosi)
- Esame audiometrico.
AMPPE (Acute multifocal placoid pigment epitheliopathy)
 I pazienti colpiti sono generalmente giovani o di mezza età (20-50 anni), senza predilezione di sesso.
I pazienti colpiti sono generalmente giovani o di mezza età (20-50 anni), senza predilezione di sesso. Le manifestazioni oculari sono precedute da prodromi virali.
Clinica
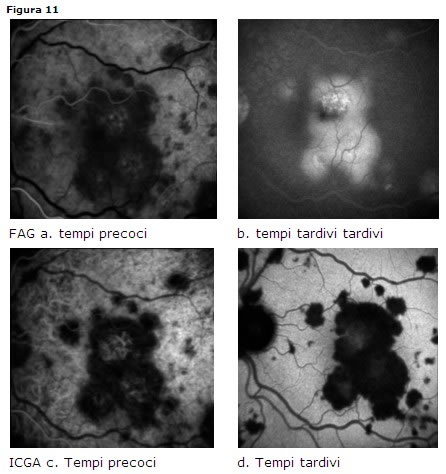
lesioni spesso bilaterali placoidi giallo-crema disseminate al polo posteriore; possono concomitare distacchi sierosi retinici. Il vitreo può essere modestamente corpuscolato.Diagnosi
- FAG: lesioni ipofluorescenti negli stadi iniziali, iperfluorescenti negli stadi tardivi (Fig. 11 a-b)
- ICGA: placche ipofluorescenti ai tempi precoci, intermedi e tardivi [41] e [42] sia nella fase attiva sia nella fase cicatriziale (Fig. 11 c-d))
- ERG e EOG: variabili [43].
MEWDS (Multiple White Dot Syndrome)
") La maggior parte dei pazienti è di sesso femminile, con unetà media di 30 anni. Talvolta la malattia si associa a prodromi virali.
La maggior parte dei pazienti è di sesso femminile, con unetà media di 30 anni. Talvolta la malattia si associa a prodromi virali.Clinica
monolaterale 90%, cellularità vitreale modesta, lesioni molto piccole bianche granulari al polo posteriore interessanti la retina profonda e lEPR.Diagnosi
- ERG e EOG: anomali
- FAG: spots iperfluorescenti precoci e tardivi, leakage disco ottico
- ICGA: caratteristica ipofluorescenza precoce e tardiva (Fig. 12)
Coroidite serpiginosa o a carta geografica
I pazienti colpiti sono adulti di età compresa tra i 40 e 50 anni. Non cè predilezione di sesso.Clinica
presentazione bilaterale, coinvolge coriocapillare e EPR, con lieve cellularità vitreale; le lesioni grigiastre ed edematose partono dal nervo ottico con andamento centrifugo, pseudopodale, in tutte le direzioni ed esitano in cicatrici atrofiche corioretiniche.Diagnosi
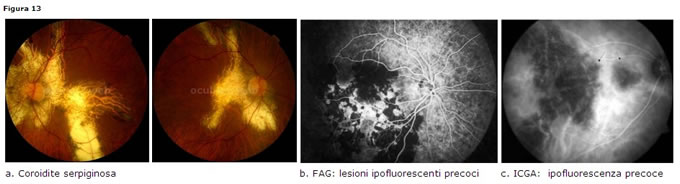
- FAG: ipofluorescenza precoce, con iperfluorescenza tardiva ai bordi della lesione (staining) (Fig. 13 b)
- ICGA: ipofluorescenza lungo tutte le fasi dellesame (Fig. 13 c)
PIC (Punctate inner choroidopathy)
Interessa giovani donne sane, solitamente miopi.Clinica:
bilaterale, assente cellularità vitreale, piccole opacità giallastre al polo posteriore localizzate al di sotto dellEPR, con evoluzione cicatriziale.Diagnosi
- FAG: ipofluorescenza precoce, con aumento della fluorescenza nella fasi tardive
- ICGA: spots ipofluorescenti.
Coroidite multifocale
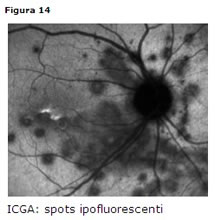 Colpisce donne giovani (età media 30 anni) e solitamente miopi.
Colpisce donne giovani (età media 30 anni) e solitamente miopi.
Clinica
bilaterale 80%, cellularità vitreale costante, numerosi spots gialli o grigi in regione peripapillare o in media periferia. Tali lesioni esitano in cicatrici corioretiniche.Diagnosi
- ERG e EOG: nella norma
- FAG: iperfluorescenza precoce dei bordi della lesione, staining tardivo della parte centrale
- ICGA: spots ipofluorescenti (Fig. 14)
Neuroretinite diffusa subacuta monolaterale
 Consegue ad infestazione da parte di nematodi ed è diffusa nel Sud-Est degli Stati Uniti, nei Caraibi e in America latina.
Consegue ad infestazione da parte di nematodi ed è diffusa nel Sud-Est degli Stati Uniti, nei Caraibi e in America latina.Clinica
essudazione vitreale, piccoli focolai retinici profondi e periferici (Fig. 15), edema papillare.Diagnosi:
- FAG: spots ipofluorescenti, leakage nervo ottico, staining tardivo
- ERG: anomalo
Epitelite retinica acuta
 Colpisce solitamente giovani in età compresa tra 20-50 anni, in buona salute.
Colpisce solitamente giovani in età compresa tra 20-50 anni, in buona salute.Clinica
piccole chiazze grigio scuro maculari; si può associare una modesta vitreite.Diagnosi
- FAG: spots ipofluorescenti circondati da alone iperfluorescente
- ERG normale, EOG anomalo nella fase acuta
4. Neuroretiniti
Le neuroretiniti sono un gruppo eterogeneo di malattie che colpisce solitamente individui giovani e di mezza età (6-50 anni), senza distinzione di sesso. Le neuroretiniti possono essere di tipo idiopatico o di natura infettiva o infiammatoria (neuroretiniti propriamente dette).La manifestazione clinica tipica della neuroretinite, bilaterale nel 5-33% dei casi, è rappresentata dalledema della papilla tipicamente precoce associato a coinvolgimento neuroretinico (stella maculare) che fa la sua comparsa anche a distanza di 1-2 settimane e che consegue non ad un processo infiammatorio retinico ma a deposito di materiale lipidico lungo le fibre nervose (Fig. 16a). Nel 90% dei casi è presente cellularità vitreale.
Laspetto fluorangiografico è rappresentato da iperfluorescenza del disco ottico, dovuta a staining, in assenza di leakage maculare.
Le cause principali di neuroretinite sono:
- Idiopatiche
- Infettive (virali, toxoplasmosi, toxocariasi, tubercolosi, sifilide, m. di Lyme, m. da graffio di gatto) [44] [45].
- Infiammatorie (sarcoidosi)
Dopo unattenta valutazione degli aspetti clinico-anamnestici generali e dopo aver escluso altre possibili cause di edema del nervo ottico con stella maculare (malattie vascolari e tumori della testa del nervo ottico) gli esami da richiedere sono:
- VDRL, FTA-ABS
- Sierologia Lyme
- Sierologia Bartonella
- Sierologia Toxocara
- PPD
- ACE, lisozima, Rx torace
- FAG
VASCULITI
Si intende per vasculite qualunque processo infiammatorio che coinvolga la parete di un vaso sanguigno che spesso, ma non sempre, porta ad occlusione del segmento interessato con possibile necrosi successiva sia del vaso sia dei tessuti dipendenti da quel vaso.La vasculite può essere uninfiammazione retinica vascolare primaria o, come avviene più comunemente, un interessamento vascolare secondario ad uninfiammazione intraoculare. Linfiammazione può coinvolgere le arterie retiniche, le vene o i capillari anche se il coinvolgimento venoso è quello più frequentemente riscontrato.
Le patologie che si presentano con un interessamento delle arterie sono la poliarterite nodosa, il LES e la necrosi retinica acuta, mentre le flebiti sono più spesso associate alla malattia di Behçet, la tubercolosi, la sarcoidosi, la sclerosi multipla, la pars planite, la malattia di Eales e le infezioni HIV correlate. Le vasculiti retiniche possono associarsi a malattie sistemiche (sarcoidosi, Behçet, poliartrite) oppure a malattie limitate al distretto oculare.
Alcuni casi invece esulano da questa classificazione e si parla di sindromi oculari idiopatiche che non hanno apparenti correlazioni con altre patologie e che vengono definite come vasculiti retiniche primitive come ad esempio la malattia di Eales [46] e la vasculite a ramo ghiacciato.
Metodologia diagnostica nelle vasculiti
Nella valutazione di questo complesso gruppo di malattie è importante la raccolta dei dati anamnestici ed un accurato esame clinico generale del paziente nonostante la maggior parte dei pazienti con vasculite, al momento della diagnosi, non presenti segni di malattia sistemica [48].E necessario riconoscere cause di tipo non infiammatorio che possono presentarsi con quadri simili a vasculite, come la retinopatia diabetica, la retinopatia da radiazioni, la retinopatia da emoglobinopatie, le occlusioni retiniche, la malattia di Coats e la leucemia.
A questo proposito è sicuramente utile la FAG che aiuta non solo ad identificare la presenza di una vasculite attiva ma permette anche di classificarla in occlusiva e non occlusiva.
Di seguito riportiamo una flow-chart utile nella diagnosi di vasculite e gli esami che è opportuno richiedere sulla base del sospetto clinico:
- Emocromo, esame delle urine
- CMV: test HIV, ELISA, PCR
- Toxoplasmosi: toxotest
- Candida: VTK diagnostica
- M. di Behcet: HLA-B51
- HZV/HSV: PCR dallumor acqueo/vitreo
- Sifilide: anamnesi, VDRL, FTA-ABS
- Linfoma: RMN orbite + encefalo, VTK diagnostica, saggio IL-10, identificazione del gene del linfoma.
- LES: ANA, anti-ds DNA, anti Sm, frazioni del complemento, anticorpi-antifosfolipidi
- PAN: ANCA
- Granulatosi di Wegener: c-ANCA, p-ANCA
- Sarcoidosi: ACE e lisozima
- Arterite a cellule giganti: biopsia arteria temporale
- Sclerosi multipla: work-up neurologico
- M. di Lyme: sierologia.
- Malattia di Whipple: PCR per ricerca bacillo, visita internistica
- Malattia di Crohn: visita gastrointestinale
- Artrite reumatoide: FR, ANA
- Polimiosite: ANA, anti-Jo1, anti-SRP
Toxoplasmosi
Descrizione
Infezione (4 tipi) causata dal protozoo Toxoplasma gondii.Toxoplasmosi congenita
Infezione acuta materna trasmessa al feto durante la gestazione; spesso asintomatica; il danno fetale è maggiore per le infezioni contratte nel primo trimestre.Toxoplasmosi oculare
causa importante di corioretinite; di solito è conseguente a un'infezione congenita ma resta asintomatica fino alla seconda o terza decade di vita.Toxoplasmosi acuta in soggetto immunologicamente sano
infezione autolimitantesi asintomatica o lievemente sintomatica.
Toxoplasmosi acuta in soggetto immunologicamente compromesso
infezione primaria o recidivante che può perdurare per tutta la vita e interessare molti organi e apparati (cuore, polmone, fegato) ma in particolare il SNC.
Sistemi/apparati interessati:
nervoso, cardiovascolare, polmonare, gastrointestinale, cutaneo
Genetica:
nessuna
Incidenza/prevalenza in USA
Toxoplasmosi acuta in soggetto immunologicamente compromesso
infezione primaria o recidivante che può perdurare per tutta la vita e interessare molti organi e apparati (cuore, polmone, fegato) ma in particolare il SNC.
Sistemi/apparati interessati:
nervoso, cardiovascolare, polmonare, gastrointestinale, cutaneo
Genetica:
nessuna
Incidenza/prevalenza in USA
Sistemi/apparati interessati:
nervoso, cardiovascolare, polmonare, gastrointestinale, cutaneoGenetica:
nessunaIncidenza/prevalenza in USA
Età predominante: tutte
Sesso predominante: maschi = femmine.
- Più del 70% degli adulti sani è sieropositivo
- Ogni anno sono affetti da toxoplasmosi più di 3500 neonati
Segni e sintomi Toxoplasmosi congenita
- Più grave se l'infezione materna avviene precocemente in gravidanza
- Assenza di segni e sintomi di infezione nel 67% dei casi
- Corioretinite (15%)
- Calcificazioni intracraniche (10%)
- Pleiocitosi ed elevata quota proteica nel liquido cefalorachidiano
- Anemia, piastrinopenia e ittero alla nascita
- Microcefalia
- I neonati che sopravvivono possono mostrare ritardo mentale, convulsioni, difetti visivi, spasticità e altre gravi sequele neurologiche
Toxoplasmosi oculare
- Corioretinite
- Lesioni a fiocchi cotonosi giallastri, rilevati, a margini indistinti
- Possibili piccoli raggruppamenti delle lesioni
- La forma congenita è solitamente bilaterale
- La forma acquisita è solitamente monolaterale
- I sintomi comprendono visione offuscata, scotomi, dolore e fotofobia
Toxoplasmosi acuta in soggetto immunologicamente sano
- 80-90% dei casi è asintomatico
- Linfoadenopatia laterocervicale lieve-media (linfonodi < 3 cm), non dolente
- Febbre, malessere, sudorazione notturna, mialgie
- Faringodinia
- Rash maculopapulare
- Talvolta linfoadenopatia retroperitoneale e mesenterica con dolore addominale
- Corioretinite
Toxoplasmosi acuta in soggetto immunologicamente compromesso
- L'infezione può essere nuova o riattivata
- Affezione a carico del SNC nel 50% dei casi
- Encefalite, meningoencefalite o masse
- Emiparesi, convulsioni, variazioni dello stato psichico
- Modificazioni del visus
- Possibile comparsa degli stessi segni e sintomi del paziente immunocompetente
- Miocardite e polmonite
Cause
- L'agente eziologico di tutte le forme cliniche è il Toxoplasma gondii
- La forma congenita è conseguente all'infezione transplacentare materno-fetale
- Le altre forme possono essere la conseguenza di una nuova infezione o della riattivazione di un'infezione latente
- L'infezione umana avviene con l'ingestione di cibi contenenti le cisti o le oocisti presenti delle feci dei gatti
- L'infezione può essere trasmessa mediante trasfusioni o trapianti d'organo
Fattori di rischio
- Stati di immunodeficienza, specie dell'immunità cellulomediata come nell'AIDS
- Il rischio della trasmissione transplacentare è maggiore durante il terzo trimestre
Diagnosi Differenziale
Toxoplasmosi congenita: altre sindromi correlate al complesso TORCH (rosolia, Cytomegalovirus, herpes simplex), sifilide, listeriosi, altre encefalopatie infettive, eritroblastosi fetale, sepsiToxoplasmosi oculare: TBC, sifilide, lebbra, istoplasmosi
Toxoplasmosi acuta in soggetto immunologicamente sano o compromesso: innanzitutto escludere un linfoma; mononucleosi infettiva, Cytomegalovirus, malattia da graffio di gatto, sarcoidosi, TBC, tularemia, metastasi tumorali, leucemia
Encefalite da toxoplasma: TBC, micosi, vasculite, leucoencefalopatia multifocale progressiva, ascesso cerebrale, neoplasia, encefalite erpetica
Esami di laboratorio
La dimostrazione del toxoplasma nel sangue, nei liquidi organici o nei tessuti è segno di infezioneL'isolamento del toxoplasma dalla placenta è diagnostico di infezione congenita
Il reperto di antigeni toxoplasmatici nel sangue o nei liquidi corporei mediante tecniche ELISA è indicativo di infezione acuta
Dye-test di Sabin-Feldman: è un test di neutralizzazione sensibile e specifico, evidenzia anticorpi IgG strettamente specifici per T. gondii, costituisce il test standard di riferimento per la toxoplasmosi, ma è un test complesso, richiede la disponibilità di toxoplasmi vivi e non è disponibile presso tutti i laboratori. Titoli elevati sono indicativi di malattia acuta
Test IFA (Indirect Fluorescent Antibody): evidenzia gli stessi anticorpi del Dye-test; i titoli dei due test sono praticamente paralleli
Test immunoenzimatici per IgM: evidenziano la presenza di anticorpi IgM nella prima settimana di infezione, il cui titolo decresce peraltro nel giro di pochi mesi
Test di emoagglutinazione indiretta (IHA): evidenziano anticorpi diversi dal Dye-test; i titoli tendono a essere più elevati e a permanere alti più a lungo
Test ELISA "a doppio sandwich" (DS-IgM-ELISA): è più sensibile e specifico degli altri test per IgM
Farmaci che possono alterare i risultati degli esami di laboratorio: nessuno
Condizioni che possono alterare i risultati degli esami di laboratorio :
Gli anticorpi antinucleo e il fattore reumatoide possono dare test sierologici falsi positivi
La gravidanza può causare test di emoagglutinazione falsamente negativi
Reperti patologici
I linfonodi mostrano tre elementiIperplasia follicolare reattiva
Ammassi irregolari di istiociti epitelioidi che invadono i margini dei centri germinativi
Distensione focale dei seni con cellule monocitoidi
Test speciali
Disponibile come test di screening un un test cutaneo d'ipersensibilità ritardata (intradermoreazione alla toxoplasmina)I livelli anticorpali nell'umor acqueo e nel liquido cefalorachidiano possono essere indicativi della produzione locale di anticorpi e quindi dell'infezione in tali sedi
Amniocentesi alla 20a-24a settimana in caso di sospetta malattia congenita
Diagnostica per immagini
TAC encefalica nei casi di toxoplasmosi cerebraleEcografia fetale alla 20a-24a settimana
Procedure diagnostiche
Biopsia linfonodaleBiopsia cerebrale nelle forme neurologiche, con dimostrazione del Toxoplasma mediante tecniche di immunoperossidasi
Trattamento
Sede di cura appropriata
Ambulatoriale nei casi di toxoplasmosi oculare e acquisita in pazienti immunologicamente saniRicovero nelle forme con interessamento del SNC e nelle forme acute in pazienti immunocompromessi
Misure generali
Di solito nessun trattamento nei soggetti asintomatici tranne che nei bambini sotto i 5 anniI soggetti sintomatici vanno trattati fino ad acquisizione dell'immunità
Attività fisica
Condizionata dalla gravità della malattia e dagli organi interessatiAlimentazione
LiberaEducazione del paziente
La madre infetta deve essere dettagliatamente informata sulle possibili conseguenze per il fetoFornire spiegazioni sulla prevenzione (per esempio, evitare che i bambini giochino dove i gatti dormono)
TERAPIA MEDICA
Farmaci di scelta
Toxoplasmosi acuta in pazienti immunodeficienti (Sulfadiazina (galenico) 100 mg/kg/die fino a 8 g/die + pirimetamina 200 mg il 1° giorno, poi 25-50 mg/die + acido folinico 10 mg/die per diversi mesi)Toxoplasmosi oculare: stessa terapia per 1-2 mesi
Toxoplasmosi acuta in gravidanza: si può prescrivere la stessa terapia dopo la 16a settimana
Toxoplasmosi congenita: sulfadiazina (galenico) 100 mg/kg/die fino a 8 g/die + pirimetamina 1 mg/kg a giorni alterni + acido folinico 5 mg a giorni alterni
CONTROINDICAZIONI
La pirimetamina non va impiegata nel primo trimestre di gravidanzaIpersensibilità nota alla pirimetamina o alla sulfadiazina (Nota: molti soggetti HIV-positivi presentano ipersensibilità ai sulfamidici)
PRECAUZIONI
La mielotossicità rappresenta un problema importante in corso di trattamento della toxoplasmosiCautela nei pazienti con possibile deficit di folati
Cautela nei pazienti con insufficienza epatica o renale
I sulfamidici possono aumentare l'effetto degli anticoagulanti orali
I sulfamidici possono aumentare i livelli della fenitoina sodica
I sulfamidici possono aumentare l'effetto degli ipoglicemizzanti orali
Raccomandare di bere in abbondanza, in quanto i sulfamidici sono poco solubili in acqua e possono precipitare nelle urine
Interazioni: i sulfamidici possono interagire con la fenitoina sodica, ipoglicemizzanti orali e anticoagulanti orali.
Farmaci alternativi
In gravidanza: spiramicina 3 g/die per 3 settimane, 2 settimane di intervallo e ripetere i cicli di 5 settimane per tutta la durata della gravidanzaClindamicina: 900-1200 mg ogni 8 ore per via endovenosa nelle forme oculari e del SNC, da sola o in combinazione con pirimetamina; può essere efficace come l'associazione sulfadiazina-pirimetamina, ma con meno effetti indesiderati
Corticosteroidi: il prednisone 1-2 mg/kg/die può essere associato in caso di corioretinite o di infezioni del SNC
Azitromicina e claritromicina hanno dato buoni risultati nella forma nervosa
FOLLOW-UP
Monitoraggio del paziente
Visite di controllo ogni 2 settimane fino a stabilizzazione, poi mensili durante il trattamentoEsame emocromocitometrico settimanale nel primo mese, poi quindicinale
Controllo della funzionalità epatica e renale mensilmente
Prevenzione
Le misure preventive sono importanti nelle gravide sieronegative e nei pazienti immunocompromessi; non mangiare le carni crude, il latte non pastorizzato, le uova crude; evitare il contatto con le feci dei gatti.Complicanze
Convulsioni o deficit neurologici focali nella toxoplasmosi del SNCCecità parziale o totale nelle forme oculari
Complicanze multiple in caso di toxoplasmosi congenita, compresi ritardo mentale, convulsioni, sordità e cecità
Decorso atteso e prognosi
I pazienti immunocompromessi sovente ricadono in caso di interruzione del trattamentoLa terapia può prevenire lo sviluppo di sequele indesiderate nei bambini sintomatici e asintomatici con toxoplasmosi congenita
MISCELLANEA
Condizioni associate
I pazienti con deficit dell'immunità cellulomediata, specie quelli con AIDS, hanno maggiore probabilità di contrarre la toxoplasmosi.Fattori correlati all'età
Pediatrica: i neonati affetti da toxoplasmosi congenita acuta spesso muoiono nel primo mese di vita; la forma subacuta può passare inosservata per qualche tempo dopo la nascita, allorché i sintomi compaionoGeriatrica: infezione acquisita; più spesso riacutizzazioni.
Gravidanza
Il rischio della trasmissione transplacentare è maggiore gli ultimi mesi della gravidanza, mentre la gravità delle lesioni è tanto maggiore quanto più l'infezione è precoceRichiedere i test di controllo per toxoplasmosi (vedi); le gravide sieronegative devono essere particolarmente attente a evitare contatti con i gatti, a non mangiare carni crude e a lavare accuratamente frutta e verdura
In caso di toxoplasmosi in gravidanza, ricorrere a consulenza specialistica